

Introduction of Mucopolysaccharidosis
Mucopolysaccharidosis (MPS) is a group of complex, progressive lysosomal storage diseases that affect multiple systems. It is caused by a deficiency of enzymes responsible for degrading glycosaminoglycans (GAGs). Mucopolysaccharides are a type of complex linear polysaccharides that are normally decomposed and metabolized within cells. Mucopolysaccharides that cannot be completely degraded will accumulate in lysosomes, which can cause developmental delays, skeletal deformities, facial appearance abnormalities, neurological involvement, cardiac lesions, corneal opacity, and other disease phenotypes.
Mucopolysaccharidosis can be classified into 7 major types and 11 subtypes based on different lysosomal enzymes encoded by different genes[1] (Table 1). Except for MPS II, which is X-linked, the rest are inherited in an autosomal recessive manner.
| MPS
type |
Subtype | Enzyme deficient | Accumulating substance | Gene involved | Inheritance pattern | MIM | Age of onset | Incidence |
| MPS Ⅰ | ⅠH | α-L-iduronidase | DS, HS | IDUA (4p16.3) | AR | 607014 | 1-2
years |
1/500,000
-1/150,000 |
| ⅠS | 607016 | 5-13
years |
1/100,000 | |||||
| ⅠH/ⅠS | 607015 | 3-7
years |
1/500,000-1/150,000 | |||||
| MPS Ⅱ | Iduronate-2-sulfatase | DS, HS | IDS (Xq28) | XR | 309900 | 1st decade | 1/150,000-1/100,000male births | |
| MPS Ⅲ | ⅢA | Heparan-N- sulfatase | HS | SGSH (17q25.3) | AR | 252900 | 2-6 years | 1/70,000 live births |
| ⅢB | α- N-acetyl glucosaminidase | HS | NAGLU (17q21) | AR | 252920 | 2-6 years | ||
| ⅢC | Acetyl-Co-A-α- glucosaminide | HS | HGSNAT (8p11.1) | AR | 252930 | 2-6 years | ||
| ⅢD | N- acetylglucosamine- 6- sulfatase | HS | GNS (12p14) | AR | 252940 | 2-6 years | ||
| MPS Ⅳ | ⅣA | N- acetylglucosamine- 6- sulfatase | KS, CS | GALNS (16q24.3) | AR | 253000 | After 1 year | 1/200,000 live births |
| ⅣB | β- galactosidase | KS | GLBI (3p21.33) | AR | 253010 | After 1 year | ||
| MPS Ⅵ | N- acetylglucosamine- 4- sulfatase | DS, CS | ARSB (5q11-q13) | AR | 253200 | 2 years | 1/600,000-1/250,000live births | |
| MPS Ⅶ | β- glucuronidase | HS, DS | GUSB (7q21.11) | AR | 253222 | Variable | 1/250,000 live births | |
| MPS Ⅸ | Hyaluronidase | HA | HYAL1 (3p21.3- p21.2) | AR | 601492 | Unknown
/rare |
Unknown |
HS, Heparan sulphate;DS, Dermatan sulphate;CS, Chondroitin sulphate;KS, Keratan Sulfate,;HAhyaluronic acid;AR,Autosomal recessive;XR, X-linked recessive.
Table 1 MPS Subtypes
Therapeutic development for MPS
The current treatment methods for MPS patients include enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), substrate reduction therapy (SRT), and various surgical procedures. The main commercially available drugs abroad are enzyme replacement therapy drugs, such as Aldurazyme (MPS I), hunterase and elaprase (MPS II), vimizim (MPS IVA), naglazyme (MPS VI), and mepsevii (MPS VII). However, the above therapies are designed to reduce non-neurological symptoms and pain and are not a complete cure. In recent years, gene therapy has been gaining momentum, aiming to achieve a one-time permanent treatment by repairing enzyme defects. Over the past thirty years, emerging research directions and treatment methods for MPS have mainly focused on gene therapy. The preclinical research on gene therapy for MPS has been increasing year by year, and clinical trials of gene therapy for certain types of MPS are underway in the United States, Australia, and some European countries[2-3].
GemPharmatech’s MPS mouse models
At present, the development of new drugs for MPS still has enormous potential and relies heavily on the support of suitable animal models. Targeting the pathogenic mechanisms of different subtypes of MPS and their corresponding defective genes, GemPharmatech has constructed numerous MPS mouse models using CRISPR/Cas9 technology.
| Strain number | Strain name | MPS subtype |
| T013936 | Idua-KO | MPS Ⅰ |
| T011969 | Ids-KO | MPS Ⅱ |
| T030220 | Galns-KO | MPS ⅣA |
| T033136 | Glb1-KO | MPS ⅣB |
| T028778 | Naglu-KO | MPS ⅢB |
| T044283 | Hgsnat-KO | MPS ⅢC |
| T035799 | Gns-KO | MPS ⅢD |
| T012455 | Arsb-KO | MPS Ⅵ |
| T023773 | Gusb-flox | MPS Ⅶ |
| T043993 | Sgsh-KO | MPS ⅢA |
Table 2 List of MPS mouse models
Data display of some mouse models:

Fig.1 The activity of IDUA enzyme showed a significant decrease in the serum of NCG-Idua-W392X mice.

Fig.2 The activity of IDUA enzyme showed a significant decrease in the liver and heart tissue of NCG-Idua-W392X mice.

Fig. 3 The GAGs content in the urine of NCG-Idua-W392X mice was higher than that in NCG control mice.
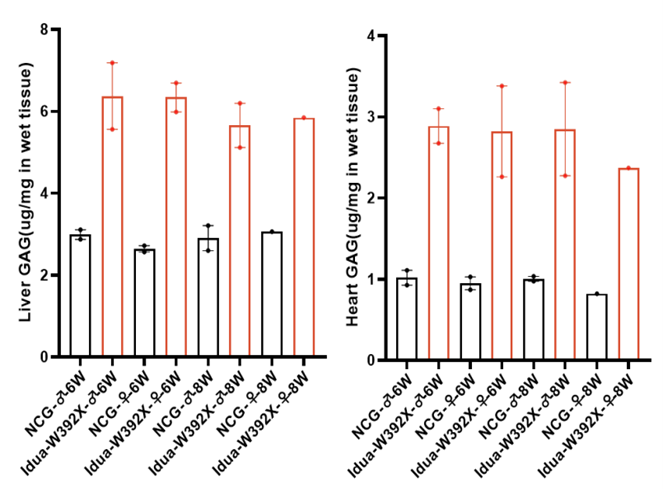
Fig.4 The GAGs content in the liver and heart tissue of NCG-Idua-W392X mice was higher than that in NCG control mice.
Reference
[1] Bhakthaganesh K, et al. Mucopolysaccharidosis. Taiwan J Ophthalmol. 2023;13(4):443-450.
[2] Ago Y, et al. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int J Mol Sci. 2024;25(2):1113.
[3] Sawamoto K, et al. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018;123(2):59-68.